Flow cytometry
Flow cytometry (abbreviated: FCM) is a technique for counting and examining microscopic particles, such as cells and chromosomes, by suspending them in a stream of fluid and passing them by an electronic detection apparatus. It allows simultaneous multiparametric analysis of the physical and/or chemical characteristics of up to thousands of particles per second. Flow cytometry is routinely used in the diagnosis of health disorders, especially blood cancers, but has many other applications in both research and clinical practice. A common variation is to physically sort particles based on their properties, so as to purify populations of interest.
Contents |
History
The first impedance-based flow cytometry device, using the Coulter principle, was disclosed in U.S. Patent 2,656,508, issued in 1953, to Wallace H. Coulter. The first fluorescence-based flow cytometry device (ICP 11) was developed in 1968 by Wolfgang Göhde from the University of Münster [1] and first commercialized in 1968/69 by German developer and manufacturer Partec through Phywe AG in Göttingen. At that time, absorption methods were still widely favored by other scientists over fluorescence methods [2]. Soon after, flow cytometry instruments were developed, including the Cytofluorograph (1971) from Bio/Physics Systems Inc. (later: Ortho Diagnostics), the PAS 8000 (1973) from Partec, the first FACS instrument from Becton Dickinson (1974), the ICP 22 (1975) from Partec/Phywe and the Epics from Coulter (1977/78).
Name of the technology
The original name of the flow cytometry technology was "pulse cytophotometry" (German: Impulszytophotometrie). Only 20 years later in 1988, at the Conference of the American Engineering Foundation in Pensacola, Florida, the name was changed to "flow cytometry", a term that quickly became popular.
Principle of flow cytometry
A beam of light (usually laser light) of a single wavelength is directed onto a hydrodynamically-focused stream of fluid. A number of detectors are aimed at the point where the stream passes through the light beam: one in line with the light beam (Forward Scatter or FSC) and several perpendicular to it (Side Scatter (SSC) and one or more fluorescent detectors). Each suspended particle from 0.2 to 150 micrometers passing through the beam scatters the ray, and fluorescent chemicals found in the particle or attached to the particle may be excited into emitting light at a longer wavelength than the light source. This combination of scattered and fluorescent light is picked up by the detectors, and, by analysing fluctuations in brightness at each detector (one for each fluorescent emission peak), it is then possible to derive various types of information about the physical and chemical structure of each individual particle. FSC correlates with the cell volume and SSC depends on the inner complexity of the particle (i.e., shape of the nucleus, the amount and type of cytoplasmic granules or the membrane roughness). Some flow cytometers on the market have eliminated the need for fluorescence and use only light scatter for measurement. Other flow cytometers form images of each cell's fluorescence, scattered light, and transmitted light.

Flow cytometers
Modern flow cytometers are able to analyze several thousand particles every second, in "real time," and can actively separate and isolate particles having specified properties. A flow cytometer is similar to a microscope, except that, instead of producing an image of the cell, flow cytometry offers "high-throughput" (for a large number of cells) automated quantification of set parameters. To analyze solid tissues, a single-cell suspension must first be prepared.
A flow cytometer has 5 main components:
- a flow cell - liquid stream (sheath fluid), which carries and aligns the cells so that they pass single file through the light beam for sensing
- a measuring system - commonly used are measurement of impedance (or conductivity) and optical systems - lamps (mercury, xenon); high-power water-cooled lasers (argon, krypton, dye laser); low-power air-cooled lasers (argon (488 nm), red-HeNe (633 nm), green-HeNe, HeCd (UV)); diode lasers (blue, green, red, violet) resulting in light signals
- a detector and Analogue-to-Digital Conversion (ADC) system - which generates FSC and SSC as well as fluorescence signals from light into electrical signals that can be processed by a computer
- an amplification system - linear or logarithmic
- a computer for analysis of the signals.
The process of collecting data from samples using the flow cytometer is termed 'Acquisition'. Acquisition is mediated by a computer physically connected to the flow cytometer, and the software which handles the digital interface with the cytometer. The software is capable of adjusting parameters (i.e. voltage, compensation, etc.) for the sample being tested, and also assists in displaying initial sample information while acquiring sample data to insure that parameters are set correctly. Early flow cytometers were, in general, experimental devices, but technological advances have enabled widespread applications for use in a variety of both clinical and research purposes. Due to these developments, a considerable market for instrumentation, analysis software, as well as the reagents used in acquisition such as fluorescently-labeled antibodies has developed.
Modern instruments usually have multiple lasers and fluorescence detectors (the current record for a commercial instrument is 4 lasers and 18 fluorescence detectors). Increasing the number of lasers and detectors allows for multiple antibody labeling, and can more precisely identify a target population by their phenotypic markers. Certain instruments can even take digital images of individual cells, allowing for the analysis of fluorescent signal location within or on the surface of cell.
Data analysis
Gating
The data generated by flow-cytometers can be plotted in a single dimension, to produce a histogram, or in two-dimensional dot plots or even in three dimensions. The regions on these plots can be sequentially separated, based on fluorescence intensity, by creating a series of subset extractions, termed "gates." Specific gating protocols exist for diagnostic and clinical purposes especially in relation to hematology.
The plots are often made on logarithmic scales. Because different fluorescent dyes' emission spectra overlap,[3] signals at the detectors have to be compensated electronically as well as computationally. Data accumulated using the flow cytometer can be analyzed using software, e.g., WinMDI(deprecated)[4], Flowjo, or CellQuest Pro. Once the data is collected, there is no need to stay connected to the flow cytometer. For this reason, analysis is most often done on a separate computer. This is especially necessary in core facilities where usage of these machines is in high demand.
Computational analysis
Recent progress on automated population identification using computational methods has offered an alternative to traditional gating strategies. Automated identification systems could potentially help findings of rare and hidden populations. Representative automated methods include FLOCK in Immunology Database and Analysis Portal (ImmPort) [5], FLAME [6] in GenePattern and flowClust [7],[8],[9] in Bioconductor. Collaborative efforts have resulted in an open project called FlowCAP (Flow Cytometry: Critical Assessment of Population Identification Methods,[10]) to provide an objective way to compare and evaluate the flow cytometry data clustering methods, and also to establish guidance about appropriate use and application of these methods.
Fluorescence-activated cell sorting
Fluorescence-activated cell sorting is a specialized type of flow cytometry. It provides a method for sorting a heterogeneous mixture of biological cells into two or more containers, one cell at a time, based upon the specific light scattering and fluorescent characteristics of each cell. It is a useful scientific instrument, as it provides fast, objective and quantitative recording of fluorescent signals from individual cells as well as physical separation of cells of particular interest. The acronym FACS is trademarked and owned by Becton, Dickinson and Company[11]. While many immunologists use this term frequently for all types of sorting and non-sorting applications, it is not a generic term for flow cytometry. The first cell sorter was invented by Mack Fulwyler in 1965, using the Coulter principle, a relatively difficult technique and one no longer used in modern instruments. The technique was expanded by Len Herzenberg who was responsible for coining the term FACS. Herzenberg won the Kyoto Prize in 2006 for his work in flow cytometry.
The cell suspension is entrained in the center of a narrow, rapidly flowing stream of liquid. The flow is arranged so that there is a large separation between cells relative to their diameter. A vibrating mechanism causes the stream of cells to break into individual droplets. The system is adjusted so that there is a low probability of more than one cell per droplet. Just before the stream breaks into droplets, the flow passes through a fluorescence measuring station where the fluorescent character of interest of each cell is measured. An electrical charging ring is placed just at the point where the stream breaks into droplets. A charge is placed on the ring based on the immediately-prior fluorescence intensity measurement, and the opposite charge is trapped on the droplet as it breaks from the stream. The charged droplets then fall through an electrostatic deflection system that diverts droplets into containers based upon their charge. In some systems, the charge is applied directly to the stream, and the droplet breaking off retains charge of the same sign as the stream. The stream is then returned to neutral after the droplet breaks off.
Fluorescent labels
- Main article: Fluorophore
A wide range of fluorophores can be used as labels in flow cytometry. These each have a characteristic peak excitation and emission wavelength. Also, the emission spectra of the labels often overlap. Consequently, the combination of labels which can be used depends on the wavelength of the lamp(s) or laser(s) used to excite the fluorochromes and on the detectors available [12]
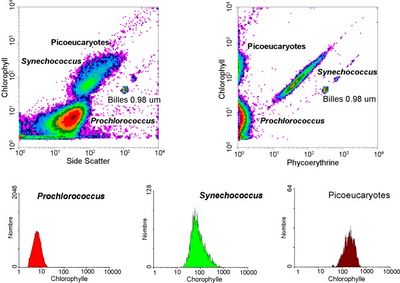
Measurable parameters
This list is very long and constantly expanding.
- volume and morphological complexity of cells
- cell pigments such as chlorophyll or phycoerythrin
- total DNA content (cell cycle analysis, cell kinetics, proliferation, etc.)
- total RNA content
- DNA copy number variation (by Flow-FISH)
- chromosome analysis and sorting (library construction, chromosome paint)
- protein expression and localization
- Protein modifications, phospho-proteins
- transgenic products in vivo, particularly the Green fluorescent protein or related fluorescent * cell surface antigens (Cluster of differentiation (CD) markers)
- intracellular antigens (various cytokines, secondary mediators, etc.)
- nuclear antigens
- enzymatic activity
- pH, intracellular ionized calcium, magnesium, membrane potential
- membrane fluidity
- apoptosis (quantification, measurement of DNA degradation, mitochondrial membrane potential, permeability changes, caspase activity)
- cell viability
- monitoring electropermeabilization of cells
- oxidative burst
- characterising multidrug resistance (MDR) in cancer cells
- glutathione
- various combinations (DNA/surface antigens, etc.)
- cell adherence (for instance pathogen-host cell adherence)
Applications
The technology has applications in a number of fields, including molecular biology, pathology, immunology, plant biology and marine biology. It has broad application in medicine (especially in transplantation, hematology, tumor immunology and chemotherapy, genetics and sperm sorting for sex preselection). In marine biology, the auto-fluorescent properties of photosynthetic plankton can be exploited by flow cytometry in order to characterise abundance and community structure. In protein engineering, flow cytometry is used in conjunction with yeast display and bacterial display to identify cell surface-displayed protein variants with desired properties. It is also used to determine ploidy of grass carp fry..
See also
- Fluorescence microscopy
Bibliography
- Flow Cytometry First Principles by Alice Longobardi Givan. ISBN 0471382248
- Practical Flow Cytometry by Howard M. Shapiro. ISBN 0471411256
- Flow Cytometry for Biotechnology by Larry A. Sklar. ISBN 0195152344
- Handbook of Flow Cytometry Methods by J. Paul Robinson, et al. ISBN 0471596345
- Current Protocols in Cytometry, Wiley-Liss Pub. ISSN 1934-9297
- Flow Cytometry in Clinical Diagnosis, v4, (Carey, McCoy, and Keren, eds), ASCP Press, 2007. ISBN 0891895485
- Ormerod, M.G. (ed.) (2000) Flow Cytometry — A practical approach. 3rd edition. Oxford University Press, Oxford, UK. ISBN 0199638241
- Ormerod, M.G. (1999) Flow Cytometry. 2nd edition. BIOS Scientific Publishers, Oxford. ISBN 185996107X
- Flow Cytometry — A basic introduction. Michael G. Ormerod, 2008. ISBN 978-0955981203
References
- ↑ DE 1815352, Wolfgang Dittrich & Wolfgang Göhde, "Flow-through Chamber for Photometers to Measure and Count Particles in a Dispersion Medium", issued 1971-01-14
- ↑ Kamentsky, Proceedings of the Conference „Cytology Automation" in Edinburgh, 1970
- ↑ http://pingu.salk.edu/flow/fluo.html
- ↑ "TSRI Cytometry Software Page". http://facs.scripps.edu/software.html. Retrieved 2009-09-03.
- ↑ "Immunology Database and Analysis Portal". https://www.immport.org/immportWeb/home/home.do?loginType=full. Retrieved 2009-09-03.
- ↑ "FLow analysis with Automated Multivariate Estimation (FLAME)". http://www.broadinstitute.org/cancer/software/genepattern/modules/FLAME/. Retrieved 2009-09-03.
- ↑ "flowClust". http://www.bioconductor.org/packages/2.5/bioc/html/flowClust.html. Retrieved 2009-09-03.
- ↑ [1]
- ↑ [2]
- ↑ "FlowCAP - Flow Cytometry: Critical Assessment of Population Identification Methods". http://flowcap.flowsite.org/. Retrieved 2009-09-03.
- ↑ "FACS MultiSET System" (PDF). Becton Dickinson. http://www.bdbiosciences.com/pdfs/brochures/23-3428-02.pdf. Retrieved 2007-02-09.
- ↑ Loken MR (1990). Immunofluorescence Techniques in Flow Cytometry and Sorting (2nd ed.). Wiley. pp. 341–53.
External links
- Flow cytometry - How does it work?
- Tutorials on fluorescence and flow cytometry
- MeSH Flow+cytometry
- Powerpoint lectures on flow cytometry
- How a flow cytometer operates
- Fluorophores.org - The database of fluorescent dyes
- Table of fluorochromes
- Java Fluorescence Spectrum Viewer
- Combined Flow Cytometric Measurement of Two Cell-Surface
- The Clinical Cytometry Society
- Clinical Flow Wiki
- High Throughput Flow Cytometry
- [3] Accessible Flow Cytometry
- FICCS - the Flow Informatics and Computation Cytometry Society
- Amnis Image Cells in Flow.
- Coulterflow Flow Cytometry Tools.
- Blue Sky Research Flow Cytometry Lasers.
- Fluorescence SpectraViewer - Check the compatibility of your fluorophores when designing multicolor experiments.
- Learn About Flow Cytometry
|
||||||||||||||